王金兰教授团队

面对种类繁多、结构丰富的材料,如何高效地筛选出具有特定性能的新材料一直是材料、物理、化学领域的难题。王金兰课题组结合人工智能技术与第一性原理高通量计算,较早开发了基于机器学习算法的单目标/多目标性质导向的功能材料筛选方法,并应用于钙钛矿光伏材料与二维铁磁材料的高效搜索,从二万余种钙钛矿和近四千中二维材料中成功筛选出数百种性能优异的太阳能电池候选材料和近百种二维铁磁半导体/半金属/金属材料。代表性工作包括Nat. Commun. (2018), Adv. Mater. (2020),Chem (2021)。该方法为新型功能材料的高效筛选和材料的逆向设计提供了切实可行的方案。由于我们在这一领域的影响,项目负责人受邀在J. Phys. Chem. Lett.上撰写前沿展望。
1. 有机无机杂化钙钛矿太阳能电池材料的高效筛选。
钙钛矿材料因其蕴含巨大复杂性和多样性的ABX3结构,具有丰富的物理性质和实际用途。近年来,以甲胺碘化铅为首的有机无机杂化钙钛矿(HOIP)材料因效率高、廉价、易制备等优点在各类太阳能电池材料中脱颖而出,但存在的环境稳定性和毒性问题制约了其在实际中的应用。针对这一问题,我们发展了以得到适合带隙为目标的机器学习算法,通过对212个已知HOIPs的训练,在第一性原理(DFT)精度上预测了5158种新HOIPs的带隙,并从中筛选出218种符合目标带隙的HOIPs。在此基础上,进一步利用DFT计算对稳定性、毒性等性质进行筛选,最后得到6种最具应用前景的太阳能电池候选材料(材料筛选流程见图1)。我们的验证结果显示HOIPs带隙的预测具有96%以上的准确率,且计算时间相较于DFT计算缩短了5-6个数量级。此外,利用特征选择算法,我们构建出了HOIPs带隙的构效关系,发现容忍因子、氧八面体因子、金属原子电负性以及有机分子极性对带隙具有重要影响。最后我们成功构建了包含1669种具有不同带隙和用途的HOIPs数据库。这一工作发表在[Nat. Commun. 2018, 9, 3405 ESI高被引],并被材料人、XMOL等特别报道。
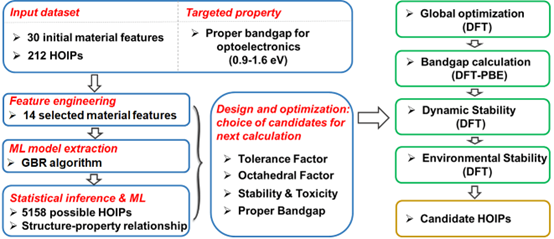
图1. 基于机器学习算法的有机无机杂化钙钛矿材料筛选流程图
值得一提的使,机器学习预测的两种化合物(CH3NH3)2CoBr4和 (CH3NH3)2MnCl4已经在实验上合成。其中,实验者利用缓慢蒸发法和反溶剂法制备了(CH3NH3)2CoBr4单晶,暴露空气24后依然能稳定存在,吸收和光致发光光谱显示其在可见光区域530nm和675nm处存在两条明显的吸收带,这和我们预测的带隙性质符合。同样的,(CH3NH3)2MnCl4单晶也通过溶剂法制备而出,相比于(CH3NH3)2CoBr4,(CH3NH3)2MnCl4在可见光区域具有更多的吸收和发射峰,实验者因此基于此材料制备出了性能优良的LED。这两种材料的实验合成证明了机器学习技术在理论指导实验方面具有较大的潜力。
2. 无机铁电光伏钙钛矿材料的快速预测。
铁电材料中体光伏效应的发现为克服无机钙钛矿带隙较宽的缺点提供了新的思路。针对寻找铁电光伏材料这一现实问题,我们结合机器学习算法和DFT高通量计算,提出了一种多目标、多步骤的材料筛选策略。针对稳定性、极化、合适带隙这三个目标性质,我们分别采用了机器学习中的分类算法和回归算法对19841种无机钙钛矿结构进行了逐层筛选,最终成功选出了151种同时满足三个目标性质的铁电光伏材料(图2),从中随机挑选出了八种进行DFT验证,机器学习预测带隙值与DFT计算值之间的误差均小于0.1 eV,且都具有自发极化。
值得一提的是,我们在特征选择方法的帮助下,首次提出了针对混X位无机钙钛矿结构稳定性的几何描述符t' 和 μ',一方面扩展了传统描述符的容忍因子t和氧八面体因子μ的适用范围,另一方面,利用它们进一步提升了预测准确率。这一结果表明,在寻找复杂体系描述符的过程中,除了依赖研究者的经验和直觉,机器学习中的特征选择算法也可以发挥重要作用。此外,在对极化结构的筛选过程中,我们通过基于DFT的小规模高通量计算来获取描述符,再通过回归算法进行大规模预测,实现了机器学习对高通量计算的加速及数据库的快速扩展。这一工作发表在[Small Methods 2019, 1900360]。
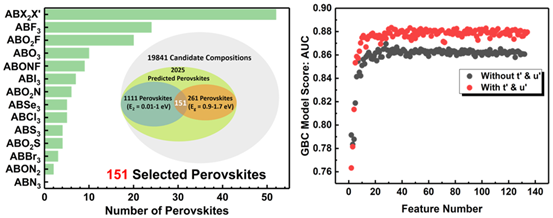
图2.多目标机器学习算法筛选得到的151种铁电光伏候选材料
3. 二维铁磁材料的高效筛选与晶体图张量描述符
二维铁磁材料在自旋电子学领域具有广泛的应用,然而大部分已知的二维材料都不存在铁磁性,同时具有半导体/半金属特性的更是寥寥无几。为了快速发现更多的铁磁材料,我们对2569种已知的二维材料进行了铁磁/反铁磁高通量计算,并结合机器学习,对材料数据库中3759种二维材料进行铁磁性预测,最终筛选出近百种稳定的二维铁磁材料。
在此过程种,由于二维材料结构复杂多样,传统的片段式描述符并不适用于二维材料。为此,我们提出了一种新型描述符——基于晶体图的三维张量描述符 (图3),该描述符不依赖于具体原子位置,只与原子近邻环境有关,并且相较于传统的一维和二维描述符,三维描述符包含了更多的物理化学信息,有利于机器学习性能的提高。我们将新型描述符应用于二维材料热力学稳定性、金属/半导体/半金属以及无磁/铁磁/反铁磁/的机器学习分类中,并通过引入反馈学习算法,实现了机器学习模型的预测准确率超过90%。最终,我们经过三次机器学习分类,筛选得到了19种铁磁半导体、19种铁磁半金属以及51种铁磁金属材料。初步建立起了包含500余种铁磁材料和900余种反铁磁材料的二维材料数据库。这一工作发表在[Adv. Mater. 2020, 32, 2002658],并被Mater View China特别报道。
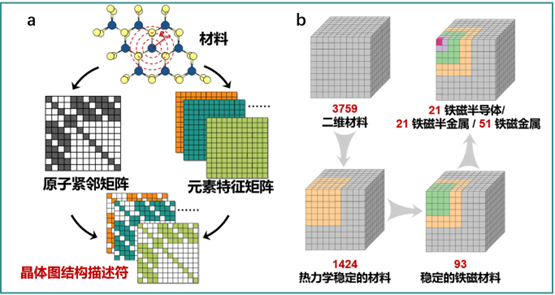
图3.基于机器学习的二维铁磁材料的高效筛选. (a) 晶体图结构描述符,(b) 机器学习筛选结果.
4. 基于主动学习的高居里温度二维铁磁材料筛选
我们在先前的工作中,结合机器学习和高通量第一性原理计算初步探索了现有的二维材料数据库中,找到了一定数量的二维铁磁体。但是实际的化学空间的规模远远超过现有的数据库,且更有可能找到性能优异的二维铁磁材料。为了搜索如此庞大的化学空间,我们提出了一个具有反馈迭代功能的主动学习框架,将梯度提升算法和边缘采样算法相结合,来动态地构建完备的高质量数据集。该框架集成了特征工程,模型学习,数据采样,第一性原理计算和模型解释功能。该框架可以使我们在数据匮乏和特征维度较高的情况下,高效地搜索化学空间。
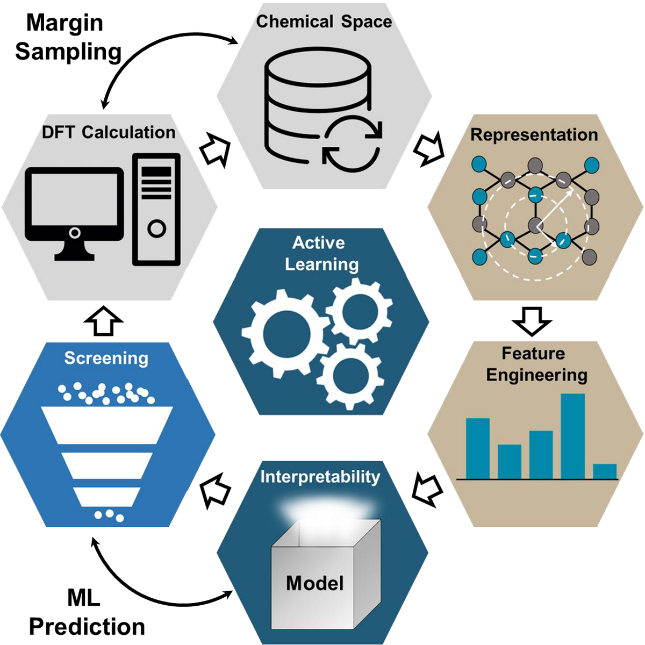
图4. 基于梯度提升算法和边缘采样算法的可解释性主动学习框架
对于主动学习而言,采样函数的选择至关重要,机器学习模型需要优先学习最能够提升当前模型性能的样本。我们在框架中采用了两步边缘采样算法,该算法在采样过程中不仅仅考虑了决策边界的样本,而且考虑了材料结构与组分的多样化。相比于随机采样,基于两步边缘采样的主动学习框架可以更快的达到模型的最优性能。
除了数据,材料描述符的构建对于机器学习模型性能的提升也是至关重要的。为了提升铁磁-反铁磁能量差回归模型性能,我们提出了一个新的局域原子化模型,改进了已有的晶体图描述符,新的描述符不仅仅包括了原子最近邻原子化环境的信息,还包括了次近邻原子化环境信息。我们将新的晶体图描述符和晶体场理论以及磁性理论相结合,最终构建出混合磁性材料描述符。该描述符只依赖于原子的相对位置和基本性质,不需要进行繁琐的计算,因此具有良好的可移植性。
我们从文献和数据库中挑选了20种具有母体材料的二维材料初始构型,构建了包含超过20万种候选化合物的化学空间。我们将主动学习框架和磁性描述符应用到该化学空间的探索中。最终,我们的模型在有限的迭代步数后,在材料热稳定性和磁性上都取得了0.9以上的预测精度。经过筛选,我们最终从化学空间中找到了9622个二维铁磁候选化合物。其中,有722个化合物的铁磁-反铁磁能量差大于0.5eV,这些材料具有较大的潜力具有高居里温度。最后,我们利用SHAP分析,揭示了磁性原子配位环境,磁性原子周期数和配位构型对铁磁性的影响。结果显示,配体场越弱,磁性原子周期数越低的四配位铁磁材料更容易出现高居里温度。这一工作发表在[Chem 2021, 8, 1]。